


Operator
Good afternoon, everyone. I want to welcome you this afternoon to Gain Therapeutics Parkinson’s Disease KOL Event “Biomarkers, Clinical Endpoints, and the Path to Disease Modification: contextualizing the emerging data from GT-02287”.
Before we begin, I would like to remind you that Gain Therapeutics management may be making forward-looking statements in our presentation today.
Please refer to this slide about forward-looking statements which describe the disclaimers and risk factors related to such statements and consult Gain Therapeutics public filings with the Securities and Exchange Commission that can be found on Gain Therapeutics website or at www.sec.gov.
Now, I’ll turn it over to Gene Mack, President and CEO of Gain Therapeutics, for opening comments.
Gene C. Mack

Thanks so much, Tara. Good afternoon and welcome. Thank you for joining us today for the Gain Therapeutics Parkinson’s KOL event. What we’d like to do here is talk about some of the biomarkers that are best understood in Parkinson’s disease and some of the clinical endpoints that will be relevant to our lead program, GT-02287, in Parkinson’s disease.
Before we get into introductions for the folks that are on the phone and joining us for the call today, we’ll just give you a brief overview. We’re going to talk about Parkinson’s disease, some general aspects of Parkinson’s disease in the context again on GT-02287.

I will describe for you a bit about a Phase 1b study Page 2 of 17 that is currently ongoing in Australia for GT-02287 in Parkinson’s patients. Then we’re going to have one of KOLs, Dr. Ken Marek, discuss the evolution of biomarkers and where we are now in terms of our understanding.
And then next, we’ll have Karl Kieburtz who will talk about the clinical endpoints from the study that we will be focusing on, particularly with emphasis in MDS-UPDRS And then we will, from there, talk through some of the preliminary or early observations we have from our Phase 1 study and then we’ll go through some of the call for some questions.
I’m joined today with our CMO, our Chief Medical Officer, Jonas Hannestad. And we are also very thrilled to be joined by Dr. Kenneth Marek and Dr. Karl Kieburtz.

Dr. Ken Marek is currently the President and Senior Scientist at the Institute for Neurodegenerative Disorders. Ken is a global leader in biomarker discovery for Parkinson’s and related neurodegenerative diseases.
He leads several major international studies, most notably the Parkinson Progression Marker Initiative, PMMI or otherwise known as PMMI, and serves as the Scientific Advisor to Michael J. Fox Foundation.
He’s also the co-founder of Molecular NeuroImaging and Xing Imaging, two companies supporting clinical neuroimaging research. And we’re also pleased to have Karl Kieburtz with us. He’s the Co-Founder of Clintrex Research and a board member at BlueRidge Life Sciences. Karl is a part time professor of neurology at the University of Rochester.
And over his career, he’s led — excuse me — he’s led more than two dozen global trials in Parkinson’s and related disorders. He’s worked closely with the FDA and NIH and several research foundations and is deeply involved in optimizing trial design and endpoint selection. Okay.
So before we dive into biomarkers, I’m going to give a broad overview of Parkinson’s disease and in the context of GT-02287. So as you may or may not know, Parkinson’s disease, the second most prevalent nondegenerative disease in the United States for sure with about 1 million patients.
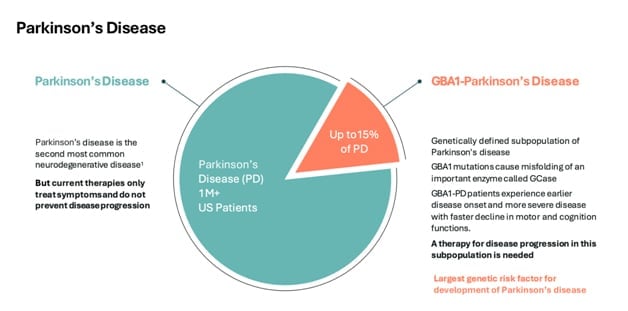
Approximately 10% to 15% of these patients have a genetic mutation in the GBA1 gene. The relevance of this GBA1 gene is that it encodes the specific enzyme target of GT-02287. So a malfunction in this genetic code creates a malfunction in this particular enzyme, and that is the targeted GT-02287.
Going forward to that reality for patients with a GBA1 mutation is that their symptoms of Parkinson’s are likely to emerge earlier in life. They tend to move faster, and it’s generally accepted to be a more aggressive form of Parkinson’s disease. So how does GT-02287 address this?
Well, in the cell, in the neuron enzyme target of GT-02287, glucocerebrosidase or GCase for short, is transcribed in the endoplasmic reticulum. From there, it has to travel to the end – to the lysosome, where it’s responsible for clearing out toxic substrates that accumulate in lysosomes as a general housekeeping function of the cell.
And from there, and more recently, we’re learning that there is an important role GCase plays in the mitochondria and support of complex one electron transport. This is an important energy support, energy support for the mitochondria, and overall mitochondrial help.
So what we believe from the preclinical data that we’ve assembled for GT-02287 is that GT-02287 engages early in the transcription and translation of GCase. It stabilizes the enzyme and chaperones that enzyme throughout its traffic pattern in the cell, helping it get to all the places it’s meant to be, all the compartments that’s meant to be in the cell with its structural integrity kept intact and for those of you who are new to enzymes, how they work, it’s a conformational shape that really converts its function so that stability initiating is very important and we believe GT-02287 stabilizes that conformational shape.
And again, I mentioned in the clinical data that we’ve assembled, that’s how we got to this hypothesis of a mechanism of action given the improvements we’ve seen in animal models of Parkinson’s disease with GT-02287 demonstrating broad neuroprotection, which is evident by its activation of GCase, the improvement in lysosomal function, mitochondrial function, a reduction of ER stress.
And these other markers also in the brain, which we will talk a lot about with the help of Ken today. And all of these aspects, all these biological markers are highly implicated in the pathophysiology of Parkinson’s disease, and we believe are addressed by GT-02287’s interaction with glucocerebrosidase.
So just a bit about the Phase 1b study. We have initiated a 90-day dosing study of GT-02287 in patients with Parkinson’s disease. Jonas will review a little bit more later on with the baseline – some of the baseline characteristics of these patients but they were more or less patients diagnosed up to seven years of Parkinson’s, stable dose of dopamine, stable dose of background medication.
We treated these patients for 90 days at 13.5 milligrams per kilogram, which is the dose level that is predicted by our animal models to gain a therapeutic index for human beings Recently we have – we have extended the open label segment to include an additional nine months of dosing.
And I’m happy to report that as of June 30, we had 16 patients enrolled. Those patients completed their 90 days. More than half of those patients have already entered the extension phase of the study with more coming, we believe 80% to 90% of the full patients enrollment from the 90-day course and we’ll roll over to the extension phase.
So we are gratified that the patients are having an experience such that they want to stay on medication. So the main objectives of the Phase 1b study, of course, are still safety and tolerability. We demonstrated this in our Healthy Volunteers study in 2024.
Additionally, we saw a target engagement in that study with an increase in GCase activity of about 53%. And we are further demonstrating endurance. We’ll talk more about the safety and tolerability profile term when we study Phase 1b, but that was the main objective here and we are very happy with the safety and the safety profile that is emerging.
We do have exploratory endpoints. Some of the reasons why we’re having this KOL call this afternoon are to discuss things like GCase activity and other biomarkers that we will be evaluating and the impact GT-02287 is having on these biomarkers.
So with that, I’m going to hand over the conversation to Dr. Ken Marek to discuss some of the biomarkers and what the thinking is around some of these things.
Dr. Ken Marek

Thank you, Gene. It’s a pleasure to be here this afternoon and have an opportunity to discuss biomarkers as they relate to Parkinson’s for a few minutes. It’s actually a very exciting moment in Parkinson’s research field because we really have been seeking to have adequate biomarkers to understand and treat and develop new therapies for Parkinson’s for many years.
We’re not quite there yet, but we are now beginning to see some light at the end of the tunnel, which I’m delighted to tell you a little bit about in the next few minutes.
So what is a biomarker? A biomarker is simply any objective measure of disease.

It’s defined very broadly and of course, we’re all familiar with biomarkers.
- Cholesterol is a biomarker
- high blood pressure is a biomarker for stroke
- PSA for prostate cancer
And of course, we in the neurology field have sought to have biomarkers that help us to define, diagnose and understand disease.
It’s a little bit more challenging because with regard to brain disorders, it’s harder to identify biomarkers and sort of access the brain. But nonetheless, there’s been enormous progress in the field of neurodegenerative diseases and more recently in Parkinson’s disease.
Why do we need biomarkers? Of course, there are many reasons.

- It’s we, you know, we need an objective measure of biology so that we can understand and track disease.
- Biomarkers may be present prior to the onset of any symptoms at all and that affords us prior to the onset of any symptoms at all and that affords us as an opportunity potentially to identify who might develop a disease and potentially prevent disease.
- There may be subsets of individuals who have specific biomarkers which can be treated and by targeted therapies.
- And, you know, I think, the ultimate reason is that for clinical drug development, we at biomarkers enable us to reduce sample size and potentially shorten the time it takes to detect a therapeutic effect.
- This is particularly important in neurodegenerative disorders, which are clinically heterogeneous and really heterogeneity is sort of the death knell to clinical trials.
And so we really want to use biomarkers to create a more homogenous sample set to be studied. When you think about a biomarker, one should think about what are you using that biomarker for?

What’s the context of use there and typically there isn’t a single biomarker for a disease, but there would be several required to potentially measure disease onset, disease progression, subtypes and response to therapy.
And of course, there are also biomarkers which are particularly focused on drug development, identifying target engagement for a drug or the pharmacokinetics or pharmacodynamics of the drug.
So as I mentioned in Parkinson’s Disease, we have had an exciting last couple of years because we have finally been able to identify a biomarker for the key underlying pathology in Parkinson’s disease. This is the aggregation of the protein called synuclein.
So this is an assay which was developed a few years ago, which is the – in which enables us to detect the aggregated synuclein Which is the – which enables us to detect aggregated synuclein, and is now widely used to help us to understand who might have the disease.
It is currently a biomarker which is only available in cerebrospinal fluid and has the limitations that it is a yes no biomarker. It’s not a quantitative biomarker, although there is a lot of energy and effort to try to improve our ability to not only detect synuclein through the CE amplification assay, but to try to quantify that as well.
Synuclein, now in association with ability to detect the downstream effects that occur in Parkinson’s on dopamine degeneration, which have been noted for again, for decades, really affords us an opportunity to develop a sort of a biomarker tandem that can define this disease biologically, which we have now sort of termed Neuronal Synuclein Disease or NSD.

We are using that term NSD for a couple of reasons. One is to emphasize that this is a biological definition. But more importantly, because we believe now this can encompass other phenotypes that arise from synucleinopathy, in particular, diffused Lewy body disease known as DLB.
So Parkinson’s DLB may have different phenotypes, but their biology is identical. And so NSD encompasses this range of disorders. So this really enables us in Parkinson’s disease to be able to now for the first time understand the disease more effectively prior to the onset of symptoms.
This is a slide which is simply meant to show the natural history of Parkinson’s disease. This bright, white area is the – is sort of the area prior to diagnosis, which was – has really been a black box.
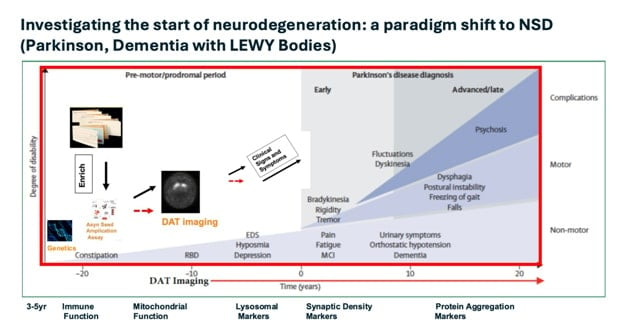
But now, as we have been able to understand and measure synuclein in life using the seed amplification assay, we can begin to sort of paint the picture of what’s happening in this white box.
So, it’s important also to know that we have this additional benefit of nature, that synuclein is highly associated with a simple test that we can use to detect individuals who might have a positive synuclein seed amplification assay, and that’s abnormal olfaction or smell.
It’s been known for years that people with Parkinson’s disease are likely to have a reduction in the sense of smell. But now, we have further associated this loss of smell with a development of synuclein, which can be easily tested with scratch and sniff tests such as illustrated in this slide.
So, the scratch and sniff test tells us who has synuclein. Ultimately, some of those people will go on to develop dopamine deficits that can be detected using brain imaging.
Ultimately, some of those people will go on to develop clinical symptoms, and this enables us now to begin to utilize this strategy to identify individuals at the earliest stages of disease more effectively.
This is just the beginning. We, as you know, recognize that there are many other pathways involved in some individuals with Parkinson’s disease, some of which are listed here. Gene’s mentioned a few as well.
So, immune function, mitochondrial function, lysosomal function, can all be affected, and we now need to be able to can all be affected or and we now need to be able to examine whether these are subsets of individuals with synuclein, whether they occur before synuclein. And these are all based on identifying appropriate, objective markers which we can detect in blood or CSF or using brain imaging to help us to better understand the onset and progression of disease.
This is all, of course, embedded in this is the importance of genetics, and certainly that’s very relevant in today’s discussion as that may well sort of be the quarterback that determines how people respond to all these biomarkers.
I’ll just mention as Gene did that I have the honor of leading the Parkinson’s Progression Marker Initiative, a study sponsored by Michael J. Fox Foundation, which has been ongoing for 15 years to identify biomarkers in Parkinson’s Disease.
And it’s this study which this commitment to this long-term effort has enabled us to validate the C amplification assay. And we are now in the process of expanding this dramatically to identify a variety of other fluid biomarkers, some of which are listed here that we think may be important in both influencing synuclein and/or perhaps determining whether synuclein is moved – goes on to develop disease in these individuals.
I’m not going to go through the details here, but just to point out that there is really a robust effort to identify biomarkers, single analytes, as well as using multiplex systems as well as using brain imaging.
Of course, it’s been a key goal in the area, in Parkinson’s Disease to identify a trace a key goal in the area – in Parkinson’s disease to identify a PET tracer that can target synuclein, just as we have PET tracers that target amyloid in tau for amyloid – for Alzheimer’s disease so that – which has really helped move that field forward.
We’re getting, I think, closer to having the synuclein trace that we seek. In fact, there are, you know, now, as you can see on this slide, several groups that have identified compounds that have the characteristics that might enable us to predict, these can be synuclein tracers, they’re not here yet, but we think 2026 may well be the year when we begin to identify tracers that can detect signal in Parkinson’s disease that would be complementary to the fluid biomarkers in Parkinson’s disease, just as they now are in Alzheimer’s disease.
We think this is a sort of a tipping point in Parkinson’s disease where we can really move the information we have of biomarkers forward more rapidly. And so, you know, we expect over the next, you know, months and years to improve our ability to define the clinical and biologic characteristics that would really now enable us to have real precision-targeted therapeutic trials.
This is – this trial we’re talking about today is an example of a precision-targeted trial focusing on genetics. But we – in addition to that, we want to be able to use the biology of disease, other key determinants and modifiers of neurodegeneration, and we want to also know how these determinants of neurodegeneration change over time so that we know what stage of disease we want to target as well.
So, what we need to do is additional biomarkers to understand both the genetics and the biochemistry and imaging digital biomarkers. I think all these are coming. And so we think this is an exciting moment in Parkinson’s.
And we’re just sort of at the start of a process. I’m going to stop there and hand it over to my colleague, Karl Kieburtz, who’s going to talk a little bit more about some of the clinical scales that we are utilizing in Parkinson’s.
Gene C. Mack
Yeah. And just thank you so much. Again, just before Karl starts to sort of remind our listeners from a timing perspective. So we will be analyzing biomarker data throughout the next couple of months and that will be available from our study later in the fourth quarter.
We’re very excited to see how GT-02287 will be impacting 00:22:27 such as alpha synuclein that will come and be available to us later in the fourth quarter.
Karl is going to review some of the clinical outward manifestations that these toxic substrates create in patients and how we are observing those changes in our Phase 1b study.
So I just want to provide some timing insight there. Thank you so much, Ken. And Karl, please go ahead.
Karl Kieburtz
Thanks, Gene. Yeah. Ken really described some of the very exciting things that are happening and being able to describe the disease processes down to the molecular level and the imaging tools that we’re starting to develop to be able to have insights into them as well as the bio fluid biomarkers.
In contrast, clinical measures may sound a little bit more antiquated. Of course, clinical measures have to do with the outward manifestations of individuals affected with these intrinsic disease processes, what we see in them as human beings, and what we can measure as other human beings. And I’ll talk about a key one of these measures.
A lot of initials up here MDS-UPDRS. MDS stands for Movement Disorder Society, which is the international society concerned primarily with Parkinson’s disease but also other movement disorders.
And UPDRS is the or the initials, the Unified Parkinson’s Disease Rating Scale. There was an initial one developed just about 40 years ago. The way rating scales were developed at that time, four guys went into a room in Bermuda and wrote up a scale about what they thought was important.
They were smart guys, they were knowledgeable, but it was very much from the expert clinician’s perspective that this UPDRS scale was developed. And it had idiosyncrasies. But it also had operating characteristics that made it useful.
And it was used for many of the approvals of the drugs we have in Parkinson’s disease. Particularly if you look back historically, parts two and three and in the old UPDRS those were assessments of activities of daily living and a motor examination. Both of those things were done by a clinician.
The new UPDRS which has only been developed this millennia is along the same lines but is different. And what you can see here is that there are four domains of it and they’re called parts, parts one through four.

And usually the first three parts are the ones that are most frequently used. And they are in this circumstance done by different kinds of raters. The first two elements of the UPDRS are actually primarily self-reported by an individual and/or their caregiver.
- And the first section is about experiences of daily living that have to do with non-motor features of Parkinson’s disease, pain, apathy, and cognitive problems. And then 13 items asking people about how those experiences impact their function.
- The second part is the same idea, experiences of daily living as reported by a person, about how things like tremor, rigidity, motor features impact their daily function.
- The third piece and the one you’ll hear the most about later from Jonas has to do with an examination done at a specific point in time by a clinician, which will rate things by their severity, things like tremor, how much a person is shaking, or how fast people can do things like tap their fingers or open their hand. So it’s an impairment scale that is done at a moment in time. I would think of that as like today’s temperature.
And the other two things are like what season it is because they reflect experiences reported by the patient over the last week whereas the examination is done at a point in time.
And in this particular study, the examination was done at a point in time taken into account when people took their last medication, if they were on medication. So if people have not taken medication overnight, let’s call in off-for-examination, meaning you’re off your medications, you’re in a practically defined, unmedicated state, or practically defined off-state.
So you’ll hear this language. And that has to do with the motor examination. What about the regulatory significance of these things? What has traction with regulators?
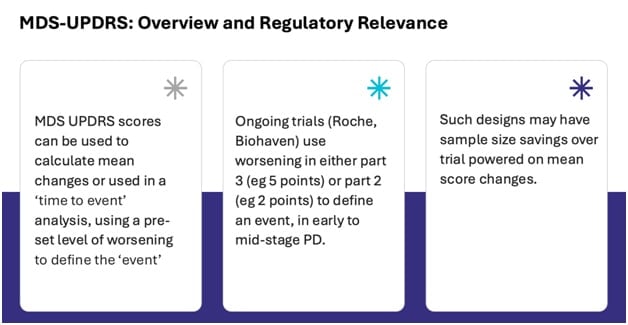
In the past part three, the examination carried a lot of weight, particularly when it was combined with part two. There’s a growing regulatory concern about the clinical meaningfulness of an examination without an understanding of the clinical impact of that impairment on a person’s function.
So both at the US, FDA and the EMA and the EU, there’s a greater reliance on the self-report of function that is part two. So usually part two is thought of as having more regulatory traction than part three.
That said, part three is the quickest way. This is the motor examination is the quickest way to see the impacts on the motor features of the disease, which are the core features of the disease.
How do we use these scores? How can they be used in the context of clinical trials?
The most traditional way is to look at mean changes. Now how bad is the impairment or the motor features at one point in time? How bad are they at a second point in time and look at the mean change.
Some of these events are pretty slow moving. Some of the changes, particularly in part two take really six to nine months to evolve in early and even mid-stage Parkinson’s disease. So seeing these changes can take quite a long time.
So alternate approaches are being thought about, including what’s here in the middle, is using a survival or time to end point kind of approach. We see this in cardiovascular trials, time to MRI or time to cardiac death or time to stroke using that kind of time to endpoint analysis has also been used.
So for example in the Roche studies has also been used, so for example, the Roche studies of prasinezumab looking at time to change of a certain magnitude in Part III and V point change which is thought to be on its phase clinically meaningful for the Biohaven’s study of their JACK – TYK2/JAK inhibitor looking at time to 2-point change in Part II, which again was thought to be on its phase clinically meaningful.
So this may improve the efficiency of trials, looking at the time to event of modicum of change in a scale which is thought to represent a significant clinical deterioration. So there are pros and cons of using the mean change versus time to event kind of approach.
But both of these are trying to identify a clinically meaningful change in the status of patients. Part II is probably the scale which on its face is most accepted by regulators, but others can be argued. There are other scales that you’ll hear about that Jonas will talk about scales of cognition, things and scales of function.
They carry a bit less regulatory weight at this point, but help describe the overall response to the intervention in this kind of patient population. So with that, I’ll hand it over to Jonas.
Jonas Hannestad

Thank you, Karl. Thank you, Ken. That’s a very, very nice overview of both of your sections. So, I want to talk about some interim results that we have from the study that Gene was describing earlier.
So this is a 90- day Phase 1b safety and tolerability study in people with Parkinson’s that’s ongoing in Australia.
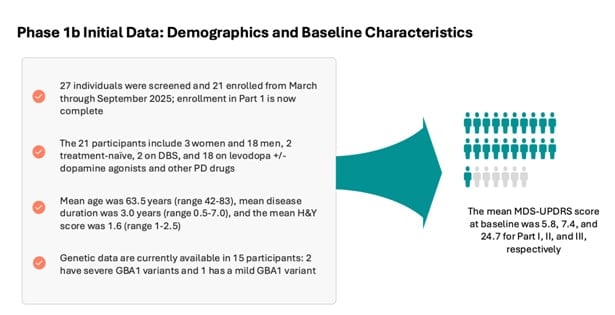
And this study had the aim of enrolling 15 to 20 participants with Parkinson’s, and we completed enrollment in early September and we have 21 participants. Completed enrollment in early September.
- We have 21 participants.
- We screened a total of 27.
And this, the 90-day part of the study, which we now call Part I, because we have the extension that Gene also mentioned, this first 90-day period will be completed by the end of this year.
So all these 21 patients will have completed their 90 days of dosing, and that’s when we will have the full dataset from this Part I. The extension will continue. So we have some patients already rolled over into the extension and we will have more in the coming months.
And because of the nine month duration, we’ll go into the first half of next year and we’ll have data from that, that part from Part II, in the second half of next year. So what do we have so far in terms of demographics?
- So we have, as I said, 21 participants, three of whom are women, 18 are men.
- And two — only two of these are treatment naive.
- The majority are on treatment. So these are — most of these patients are on levodopa.
- And some of them also take other dopaminergic or other Parkinson’s drugs.
- And then, two of them are on deep brain stimulation.
So, it’s a sort of a mixed population that’s reflective of the stage of disease.
- So these are — the mean age of these participants is about 64, three years of disease duration, the mean, we have a cutoff at seven.
- And then the mean H&Y stage which is a sort of a staging scale use in Parkinson’s is 1.6.
So these participants are fairly representative of the early to mid-stage Parkinson’s population. With regards to genetics, we have — we have some, some genotyping data still pending.
But what we know so far is that of these – we have three GBA carriers,
- Two of whom have the severe variants and
- One of them has a mild variant
So the GBA variants that also Gene was speaking about earlier depending on how they affect Gaucher disease, which work when you’re homozygous they’re classified into severe and mild. And that has appeared to have an impact in Parkinson’s, just the severe variants are associated with a faster rate of progression in Parkinson’s.
With regard to safety and tolerability, as Gene also mentioned, we did a Phase 1 study in healthy volunteers last year and this drug was overall safe and well tolerated in that study without – we dosed only for 14 days and that same here were dosing up to 90 days.

So we have more – the safety data with longer duration of dosing. What we’ve seen so far is that in the other 21 participants,
- 18 have had adverse events,
- So 93 total adverse events.
- The majority of these, about 85% were mild and then the remainder were moderate and some more severe.
The most common adverse events are listed here. So headaches, lab abnormalities, diarrhea, fatigue and nausea. And these are for the most part, transient, short lived and, as I said, mild.
So these adverse events do not lead to discontinuation except for one participant. And this was a person who hadn’t studied drugs for about a month. But then they had – they had panic attacks, they also had nausea and headaches and decided to withdraw from the study.
Now, whether this is related to the drug or not, this is hard to say. We’ve had a couple or three patients, others who had to reduce their dose. So if you remember the slide that Gene was showing earlier, there’s the option they can reduce from 13.5 milligrams per kilogram to 11 milligrams per kilogram in case there’s an issue with tolerability.
And we had three people to do that. In case there’s an issue with tolerability, and we had three people do that. So, almost due to headaches, and two did this because of lab abnormalities, which I’ll get to in a second.
And we also had some dosing interruptions, and this was discussed with the site investigators on a sort of case-by-case basis. One, interrupt the drug for about a week due to constipation.
Again, whether this is, you know, related to the drug is hard to know because it is a very common non-motor symptom in Parkinson’s patients, and we have two patients who interrupt the drug due to lab abnormality.
So, one of them had problems 00:36:02 and changes in liver enzymes, so primarily alkaline phosphatase and GGT, and the other one had challenging changes in lipase, and these were – so these – the doses were reduced in these two patients, and then when the drug was – and there’s also an interruption when the drug was reintroduced, these lab abnormalities had normalized and continued to be normal for the remainder of the 90 days.
Regarding plasma levels and plasma exposures, we see basically exactly the same that we saw in the Phase 1 study in healthy volunteers. So, in healthy volunteers, they’re predominantly younger than the typical Parkinson’s disease population.
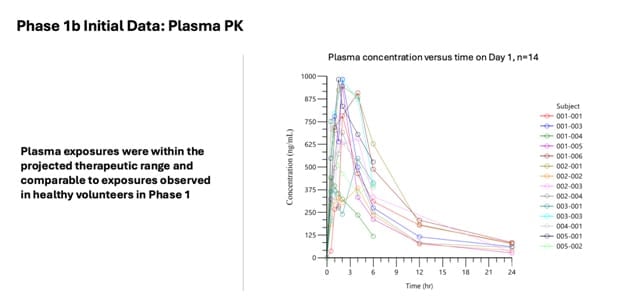
So it’s not always the case that PK is similar in younger individuals than older individuals because our metabolism changes, our liver metabolism changes. But in this case, the exposure that we see is about the CMAX, the peak plasma concentration, and the overall exposure, so the area under the curve is very similar to what we saw in healthy volunteers at the same dose level.
And the important thing is that this exposure is within the window where we think the therapeutic exposure is, and that’s, again, based on all the 00:37:26 data we have we know that in our mouse models there are certain plasma levels that are associated with a variety of improvements in brain pathology and biomarkers, et cetera and we are achieving those same exposures in these patients.
And lastly, Karl is speaking about the MDS-UPDRS, which is very helpful because here are some data on that. So again, important to keep in mind, this is an open label study so we don’t have a placebo control. So, these data have to be taken and put that caveat in mind.
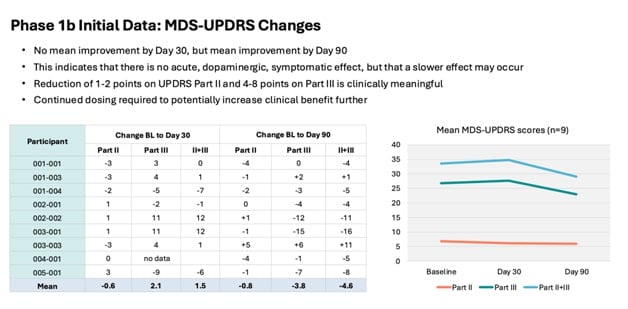
However, what is interesting is that if you look at the mean change over time, if you look at those the graphs there on the right, there seems to be – there’s no mean improvement by the 30. So there’s no sort of acute dopaminergic type effect that you would see would say levadopa.
But by the time they get to day 90, there is a mean improvement. So whether again, whether that is a real improvement in the absence of placebo, it’s very difficult to say. But that is one of the reasons we decided to extend the study because we want to characterize this so further.
So we won’t take this out from 90 days all the way out to the year to see if there’s further improvement or a sustained improvement in these UPDRS scores. And as you can see here, also, part three is the one that changes the most.
And as Karl mentioned before, that’s the one that is, you know, it changes more easily because it’s a clinician rated scale whereas part two tends to change more slowly if it changes. So, that’s also another reason to see if, you know, if this small improvement in part two Small improvement in Part 2 will continue over the course of the next nine months.
So what are the next steps in the development of the strokes?

As I said, the 90 days, the Part 1 of the study, the 90 day part will be completed by the end of this year. So we’ll have both full data sets who will have all the safety and tolerability data. We’ll have all the PK data. We’ll have all the UPDRS data.
And then most importantly is the biomarker data. So going back to what Ken was talking about before, we are making a variety of biomarkers that are associated with this mechanism. So sort of target engagement biomarkers.
And biomarkers that are associated with the disease like alpha synuclein. And those will tell us both whether the drug has its intended biological effects from what we’ve observed in both in vitro and in vivo.
And most importantly, whether the drug has an apparent effect on the biology of Parkinson’s disease. So these biomarkers will be coming up again towards the end of the year. And we are planning, we’re planning for a Phase 2 study.
So once we have this full data set a Phase 2 study that would start sometime towards the end of the second half of the first over the next year so in Q2 of next year. And this will be a placebo controlled study to really determine whether this improvement in the UPDRS scores that we see is real when you have concurrent placebo control.
And the IND for that Phase 2 study is planned to be submitted by the end of this year. And with that, I’m going to hand it over.
Gene Mack
Thanks. I just want to summarize a couple of things and some respects, especially one of the things that patients and physicians involved in the study said was not a trivial matter for them.
Some invasive sampling with lumbar puncture. So we are grateful and in their debt and are gratified that they have in a large majority chosen to stay on stay on GT-02287 through the extension phase of the study.
We will be able to gather even more important evidence, because I think, as I hope all of you have gotten a chance to appreciate, there’s — it’s a very difficult thing to impact the biology of these patients in a meaningful way, to lead to a clinical outcome and as observable within a 90-day timeframe, within the extension phase of the study will bear this out for us a bit more, and really and really show us exactly how far we can go in delaying the signs and symptoms of Parkinson’s disease.
So we’re very, very excited to have the extension phase this morning. We’re very, very excited to get the biological evidence that we’re hoping to find in these patients that have proven to be somewhat in the early days of the study. So stick with us and stay tuned.
We’re going to open up the call to Q&A. I believe we have some already cued up and I’m going to hand it over to somebody much more capable of doing that.
Operator
Yeah. We’ll do as the questions come and we will just sort of try to guide them towards the right speaker if it’s not obvious.
QUESTION AND ANSWER SECTION

Operator: Great. Thanks, Gene. So, yeah, please hold for a brief moment to pull for our questions. So our first question comes from Jay Olson at Oppenheimer. Please go ahead, Jay.
Jay Olson: Oh, hey, guys, congrats on all the progress and thank you for hosting this educational event. We had a couple questions. Are there any differences in the rate of Parkinson’s disease progression between GBA1 mutation carriers versus idiopathic patients? And I guess, is the GBA1 mutation considered a prognostic factor for Parkinson’s patients? And then we had a couple of follow ups, if we could, please.
Gene Mack: Karl, that’s probably right up your alley.
Karl Kieburtz: Yeah. And Ken may have thoughts. So, traditionally the GBA1-PD phenotype is thought to be slightly more severe than idiopathic, and have more cognitive features, and perhaps have a more rapid rate of deterioration. Looking at that data doesn’t call into question how different they are, but if anything, they’re slightly more cognitive impairment, maybe slightly more rapid progression, but it’s not notably different. I don’t know, Ken, if you feel differently than that.
Ken Marek: No, I would. I would agree with that, and also just point out that there are a number of GBA1 GBA mutations and some of which seem to be more severe than others. So, there’s a range of mutations and a range of how that influences the severity of illness. But, I agree with Karl, that typically, it’s a little bit more severe than that of a typical PD individual.
Jay Olson: Great. Thank you. Thank you for that. And then based on the mechanism of GT-02287 how long would you expect it to take to see changes in UPDRs part two or part three scores Especially since other therapies like alpha-synuclein antibodies, have taken a while to show separation fairly late in the course of treatment.
And then, depending on the changes that you do see with GT-02287 how would you distinguish between symptomatic treatment benefits versus disease modifying benefits?
Jonas Hannestad: Yeah, I can take that one. So I think those are two key questions so I think we you know, we expect our drug to be more like Roche’s you know alpha-synuclein antibody because it’s presumably our mechanism is disease modifying and we don’t see the typical acute effects that we you know, you see with the dopamine agonist for instance or levodopa.
Now the fact that we’ve seen some improvement in 90 days was somewhat surprising, you know, so we don’t really understand the mechanism behind that. But it’s not as I said before, it’s not the typical fast effect that you will see.
But levodopa you’ve seen in a matter of hours, right, and days. It’s not that effective. It’s something that takes longer that may happen inside the cells so that you have dopamine neurons that are not functioning well because of these pathway abnormalities.
And then GT-02287 corrects some of that and that over a matter of weeks enables those neurons to work better. Right? So that’s a hypothetical explanation at this point. But I think to see it really robust, we have to look at much longer studies, that’s why the next study is a year and also the reason for the extension.
I think that the question of, you know, symptomatic versus disease modifying is also tricky because what they’re trying to demonstrate here, I think, taking because what they’re trying to demonstrate here, as I think as, and then taking Karl’s words here is that you’re making patients not get worse as fast, right?
So, it’s just that you’re not expecting an improvement necessarily expecting them to over a long period of time not get worse as much as if they didn’t take the drug. And that is by the, you know, that inherent characteristic of the disease requires much longer trials.
Jay Olson: Great. Super helpful. Thanks so much for taking the questions.
Operator: Great. Thanks for the questions, Jay. Our next question comes from Tom Shrader of BTIG. Please go ahead, Tom.
Analyst:Tom Shrader
Tom Shrader: Can you hear me okay? I just.
Operator: Yes, we can.
Tom Shrader: I have, well, I have such KOLs on the line. I kind of had a remedial question on alpha-synuclein. Is it, you know, we – I think more about Alzheimer’s disease, is it close, closer to a-beta where there are all levels of pathology before you really have clinical symptoms? Or is it closer to Tau where if you’re seeing alpha-synuclein, you’re really in the steep part of the curve and it’s time to treat? Is it known at that level yet?
Ken Marek: Yeah. That’s a good – that’s a great question. And I think I can answer it on a couple of levels. I think in some ways, of course, alpha-synuclein is like a-beta in that it occurs very early and that it is possible to have a-synuclein present for many, many years without any symptoms at all.
And then ultimately it will – in some individuals lead to symptoms. On the other hand, it also has some similarities to Tau, and that unlike a-beta, which by the time symptoms arise is everywhere in the brain, synuclein is in specific As is everywhere in the brain.
Synuclein is in specific locations in the brain. And tends to spread like tau overtime causing, and at that point, causing symptoms which seem to be related to the spread of the synuclein. So I think we need to learn more about that and the biomarkers that we have now and acquiring will help us.
But I think it also raises the point that really as we think about neurodegenerative diseases, these occur, you know, often together. They have – you know their functions and their function is similar in different ways.
And I think one of the real questions is, you know, why synuclein, why e-beta occurs in individuals as they age. So that’s an important issue. And again it’s often the case that these occur as co- pathologies. And so maybe we’re thinking maybe we should be thinking about neurodegeneration as a wider – with a wider view not so much as either AD or PD.
Tom Shrader: Okay. A couple of more clinical follow ups. To follow on Jay’s first question, just from what you know about these rating scales, is there anything you would expect to change first any leading indicators? And then you flashed a little bit of data very quickly.
And I just noticed one of the patients, I think 003-001 was up 10% in the first three months and then down 15%. Is that kind of the inherent noisiness of the data? Do you really need a lot of patients to see an active drug?
If a drug is clearly active in a meaningful way how many patients do you think you need to see it I guess is the way. I guess I’d love an estimate for? Thank you.
Karl Kieburtz: I’ll jump in on the clinical scales. Are they that noisy? Yes. Especially in people who are already on dopaminergic treatment and how they respond to coming on and off. These are somewhat artificial states that we’re measuring people in, in terms of practically defined off state.
So, yes, there is a lot of nosiness in the, you know, point estimate or the point measurements at any point in time. And we do power around that. So for drugs that are clearly, you know, effective in terms of symptomatic improvement, like domain agonists, which are not as powerful as levodopa, maybe 50 groups is the size you need to show changes.
You know, levodopa, you can show with half that because it’s a monster effect. It gets a little. And that’s short-term improvement. It’s a little bit more complicated of, you know, how many do you need to show long-term, less worsening, as Jonas was saying. And there again, the variability comes into play.
But probably the effects there are going to be smaller. I mean, I’ll go to lecanemab and donanemab. These are 20% reduction in deterioration which you would hope for something more potent than that. But there are large numbers to be able to detect those kinds of differences.
Tom Shrader: And then any thoughts on what is there any one scale or something that if you saw a change, you’d say, that’s pretty meaningful?
Karl Kieburtz: No. I think, you know, parsing the subparts of these scales, either two or three, to say that’s the leading indicator from within the scale is that’s the leading indicator from within the scale was not a full’s errand but I think this can be more misleading than informational. I don’t know. Ken, you might feel differently.
Ken Marek: You know, it’s – I think it’s unclear which way – which of these – which of the parts is most effective, I think. You know, I mean, you know, typically, you see greater change in Part III than Part II.
So you can you know, so I think that’s always encouraging and gets more widely used and in that regard. But, you know, I don’t know that there is a leading indicator that’s reliable, you know.
Tom Shrader: Okay. Great. Thank you for all the details.
Operator: Thanks for the questions, Tom. Our next question comes from Boobalan Pachaiyappan at ROTH. Please go ahead, Boobalan.
Analyst: Boobalan Pachaiyappan
Boobalan Pachaiyappan: Hi. Can you hear me okay?
Operator: Yes. We can.
Boobalan Pachaiyappan: Awesome. Congratulations on the progress and thanks for taking our questions. So two from us. Firstly, talking about the treatment adherence rate, can you maybe discuss at a high level how does the treatment adherence rate in IPD patients compare to the GBA1 PD cases?
And then I see that more than 50% opted to enroll in the open-label state or open label state of the study. So assuming there’s also treatment discontinuation in the subpopulation during the course of the therapy, what is the minimal sample size that must complete the nine months of treatment in?
Jonas Hannestad: Right. So yeah. I think I had that. So, we haven’t – so we only have, you know, one, one part has been discontinued so far and that was present with idiopathic Parkinson’s, so we can’t really tell if there’s a difference in between GBA and idiopathic.
My guess is that the adverse events or the, you know, the tolerability challenges that some of the adverse events or the, you know, tolerability challenges that some of these people may experience are unrelated to the genetic background, though that would be the most likely explanation or scenario.
So in that case, the discontinuation would be sort of a, you know, a random event in which a certain person has problems tolerating more side effects, right? And we don’t know how many of the people are continuing the extension for nine months.
My guess is that if they all have tolerated the drug and some of them have improved over the course of three months, then they will be more likely to stay on. But, you know, part of that is our hope that we don’t know yet. So, we’ll see.
You know, that’s important information that we’ll get from the study as well as adherence rates and attrition.
Boobalan Pachaiyappan: All right. Great. I have one question for Karl. So how normal or abnormal is it for patients who are treated with investigational agents such as GT-02287 to show stabilization or improvements in UPDRS Part II and Part III in like 90 days?
And how is the safety efficacy profile of GT02287 compared to some of the other clinical agents that also target similar pathways of GKs? Thank you.
Ken Marek: I would love to start. So I think it’s not common to see clinical effects at 90 days. And I think as, you know, I already, I think, mentioned I think it’s hard to interpret that. That’s really why you need to really extend that period to really understand better if these changes are meaningful.
I think that the safety profile, you know, is a favorable profile so far. And, you know, of course, additional information will be acquired as the studies are expanded. But I – certainly, it’s a favorable profile with regard to the other tested drugs, you know?
Karl Kieburtz: I agree.
Boobalan Pachaiyappan: All right. Thank you.
Operator: Thanks for the question, Boobalan. Our next question comes from Chad Yahn at Maxim. Please go ahead, Chad.
Analyst: Chad Yahn
Chad Yahn: Hi, guys. Thanks. I’ll be brief. So granted, it’s still quite early, but do you think the motor improvements we’re seeing point to lysosomal restoration in dopamine genic pathways, or could they just be downstream?
Jonas Hannestad: You know, that’s the question we want to know the answer to, right? So based on what we have seen in animals, the restoration of some of these pathways is fairly fast. So we see a decrease in alpha-synuclein aggregation in the brains of these animals and so these are mice. We see an improvement in lysosomal function, improvement in mitochondrial function.
So — but you know, which one of those or which combination of those is the one that then leads to the behavioral improvements in animals. We don’t really know because it’s very difficult to correlate and the same is true in humans, right? That’s something we may, we may never have the answer to.
We may once have biomarker data. We may see that certain biomarkers change more than others. Now, we can basically hypothesize that those things are driving any clinical effects. But it’s kind of an inscrutable question.
Karl Kieburtz: I will just say that interfering potently with a biologic process, like might be happening here, does, to your point, have the possibility to relieve synaptic dysfunctions, a broad kind of category that Jonas was talking about, and the relief of a synaptic dysfunction could lead to either stabilization or actually short term improvement, neither which is a bad thing in the long run, as well as the slower deterioration, sort of stabilizing your background.
So, it may not be necessary, but it may be part of the treatment effect. I wouldn’t confuse that with being, “symptomatic.” It probably has a longer time-frame than dopaminergic drugs to see the release of relief of synaptic dysfunction and may evolve over weeks. But that could happen, if that’s what you’re getting at Chad.
Chad Yahn: Got it. Thanks, guys.
Boobalan: Thanks for the question, Chad. Our final question comes from Ram Selvaraju at H.C. Wainwright. Please go ahead, Ram.
Analyst: Ram Selvaraju
Ram Selvaraju: Thanks very much for taking my questions.
Firstly, I wanted to ask if it would be possible perhaps for the KOLs to correlate impact on GCase the way, for example, a drug like GT-02287 is purported to act, with the likelihood of impact on a particular stage of Parkinson’s disease.
So, for example, you know, would we consider a drug acting on GCase to be particularly beneficial in earlier stage or later stage patients, just based on the mechanism of action?
Secondly, I was wondering if you could perhaps comment on where a mechanism like this, an
intervention with a mechanism like this might fit within the overall Parkinson’s armamentarium, in particular, when we think about, you know, other drugs that are used to typically manage Parkinson’s disease, where this drug might fit best with what additional existing approved medications it might synergize best.
And then lastly, maybe it would be possible to comment briefly on the mode of improvement that we are seeing so far and correlate that perhaps to some of the animal evidence, some of which, if my memory serves me correctly, did indeed not only point to the potential for this compound to slow disease, but also potentially even reverse certain symptoms of disease.
And I was just wondering, you know, whether the KOLs might venture an opinion on what the likelihood is of potentially seeing some kind of symptom reversal or functional recovery as we go further into clinical investigation with the compound. Thank you.
Gene Mack: Go ahead, Ken.
Ken Marek: So, these are – those are three great questions. Now, I’ve already forgotten the first, the second one. But, you know, I think that, you know, at what stage might this be useful? I think, you know, it’s of course, hard to predict that.
But I think, you know, typically it is, you know, kind of likely that at an earlier stage of disease, before degeneration has occurred, this strategy, in fact, like other strategies, might be more useful.
That’s not to say it could not be useful later, but I think that, that would be – that would be my – what I would suspect would be the case. I think that now I have really forgotten the second question, but the last question is, you know, is, you know, really related to, could this result in a reversal of symptoms?
You know, certainly that’s possible, as Karl just mentioned Certainly, that’s possible, as Karl just mentioned, that this really does affect synaptic dysfunction. Theoretically, that would be so. My guess it would take some time for that to happen.
But I think that that would be a wonderful thing. But even the – I would just point out that even the slowing of progression or the stabilization of illness would be a remarkable effect. That would be, you know, really of great value for patients. So I think we – you know, what we’d like to see a reversal even without that, it would be, it would be very, very valuable.
Karl Kieburtz: Yeah. I think hoping for improvement might be too much. And there’s kind of a yin and yang of the impact of treating very early is going to be greater. You know, you’re going to be able to spare more neurons growing early is the optimal strategy.
But from the pragmatic point, it’s hard to measure things very early because very little is moving. So you’d rather get a little later so that you have more signal to measure that things are deteriorating or stabilizing even though there’s more neurons lost.
So there’s tension between going early and late, and you try to find the right spot in there where people are not too far gone. So you really don’t – this is probably not a drug for people who are, you know, already had, you know, major complications, they’re demented, they’re far on into their disease is probably not the right.
But everything before that, it makes sense because it could help stabilize them, potentially improve them, and try to drive towards the earliest stages to do the most preservation that’s possible. Ken Marek: Yeah, I would agree exactly with Karl.
I just might make one other point, which is that as we are moving toward an era where we may well have biomarkers that move prior to the onset of symptoms, it may open up the opportunity to really see more of a change due to the therapies, even at these earlier stages. So that’s just another option as we move forward. I think, again, I would agree that there’s – this is – this mechanism could easily work later as well.
But I think that, you know, it’s always a good idea to try to stop the process as soon as you can after, you know, someone has the biology that’s going to lead to Parkinson’s disease.
Gene Mack: And I can follow up on that a little bit by just saying that you’re coming out of the MDS conference where we just were presented. Some journalists have critical data there that there was an assumption that there is an emphasis in trying to relieve the dependency.
Remember that. And if we can affect some of that by creating a situation where patients need less dopamine over time and over duration, I think this would be a very, very successful comment unless you guys disagree.
But I took it away from the habeas corpus. And there’s a lot of emphasis on trying to bring these patients on the dopamine that they need. Okay. I believe we are out of questions and grow over time.
I want to thank everybody, particularly Dr. Marek and Dr. Kieburtz for joining us today and doing their best to give some insight on our clinical program and, of course, for yourself as well and all of your participation, time and interest is all greatly appreciated.
And we look forward to getting in front of you very, very soon with some more exciting data.




