Advancing GBA Modulator GT2287: A Promising Treatment for Parkinson’s Disease
- Details
- Written by: Dhaval Gupta
- Category: Perspectives
- Hits: 555
Advancing GBA Modulator GT2287: A Promising Treatment for Parkinson’s Disease
Professor Taylor highlights the groundbreaking work of Gain Therapeutics in developing GT2287, a GBA modulator, as a potential treatment for Parkinson's disease. She discusses the origin of GT2287, its mechanism of action, and preclinical findings demonstrating its efficacy in addressing key pathological features of Parkinson's disease. For a comprehensive understanding of the subject matter, we recommend watching the video for an in-depth analysis. Alternatively, to grasp the key themes discussed, peruse the accompanying blog for an overview.
Introduction to Gain Therapeutics' GT2287
Hartaj Singh: Great, Tiago. Thank you so much. I appreciate you going over so much in-depth material, and especially focusing on molecular biology and pathology. I think Dr Alcalay and you have brought us to a point where we can now hand it over to Gain’s own Dr Joanne Taylor. She is the Senior Vice President of Research here and she's going to discuss the recent in vivo data for GT 02287. It’s a best-in-class small molecule for the treatment of GBA Parkinson's disease and other neurodegenerative diseases. So Joanne, please take it away.
GT2287: Discovery and Clinical Development
Professor Taylor: Thank you very much, Hartaj. And thanks very much to our previous two speakers who have undoubtedly made my job a lot easier. So as Hartaj said, I'm going to introduce you to our GBA modulator, GT2287, which is currently in the clinic as a treatment for GBA1, Parkinson's disease.

So this molecule was discovered from our proprietary computational drug discovery platform. Actually, all of the molecules which were in our pipeline were discovered employing this platform. So we're delighted that GT 2287 is now in the clinic as proof of concept of the utility of this platform. And before I get going, I'd like to acknowledge the generous support of various foundations that have financially contributed to this work over the years. They include the Michael J. Fox Foundation, the Silver Star Foundation, [indiscernible] and Eurostars.
The Role of GT2287 in Parkinson’s Disease

So I don't think I need to go through this slide at all, given the two excellent previous presentations, just to reiterate that we think that restoring GCase function is expected to stop or slow the progression of Parkinson's disease. And we're hoping that that's what our candidate molecule GT 2287 is going to be able to do.
Efficacy of GT2287 in Cell-Based Experiments and Animal Models

So we believe that GT 2287 addresses the primary path of physiological effects in the disease cascade resulting from GK’s function that we've heard quite a lot about already from our previous two speakers. And in a plethora of cell-based experiments, some of them using cells derived from patients, and also animal models in which we have modelled GBA1 Parkinson's disease, we have seen that our compound can ameliorate lots of the effects of GCase dysfunction. So, for example, we've been able to see effects on endoplasmic reticulum stress, which is caused when misfolded GCase is not able to traffic through the cell to the lysosome where it needs to be to perform its function. This causes an accumulation of toxic lipids, that substrate of the enzyme as well as Alpha-synuclein accumulation, and we've also been able to show in many, many experiments that we can prevent this from happening with our compound.
The lysosomal dysfunction that ensues from all of this buildup of Alpha-synuclein and lipids, and GCase not being where it's supposed to be, we can also stop this happening with our compound and we can also stop the accumulation of mitochondrial reactive oxygen species. It's become clear recently that not only is GCase important in the lysosome, but it needs to get to the mitochondria as well where it's important in energy homeostasis. So having this functional GK seems to affect mitochondria, which we know are very, very important in neurodegenerative diseases, including Parkinson's disease.
And then in animal models, we've been able to show that our compound affects neuroinflammation, that it prevents neuronal cell death, particularly dopaminergic neuronal cell death, and it prevents the ensuing motor deficits from all this cascade of pathology. So because of this, we expect GT 2287 to have potential as a disease-modifying therapy in Parkinson's disease, as well as other neurodegenerative diseases where a deficit in GCase function has been implicated. So we think that our compound has a best-in-class profile of the GCase targeting drug candidates that we know about to date. It's orally available, it's a brain-penetrant small molecule, and it's an allosteric GCase regulator; it binds away from the active site, so it doesn't interfere with sub-straight processing. And by the mechanism that we heard about earlier, this compound chaperones GCase to the lysosome, where it needs to be to perform its function. And as I already mentioned, this compound is already in Phase 1 clinical trials.
GT2287’s Advantages and Allosteric Regulation
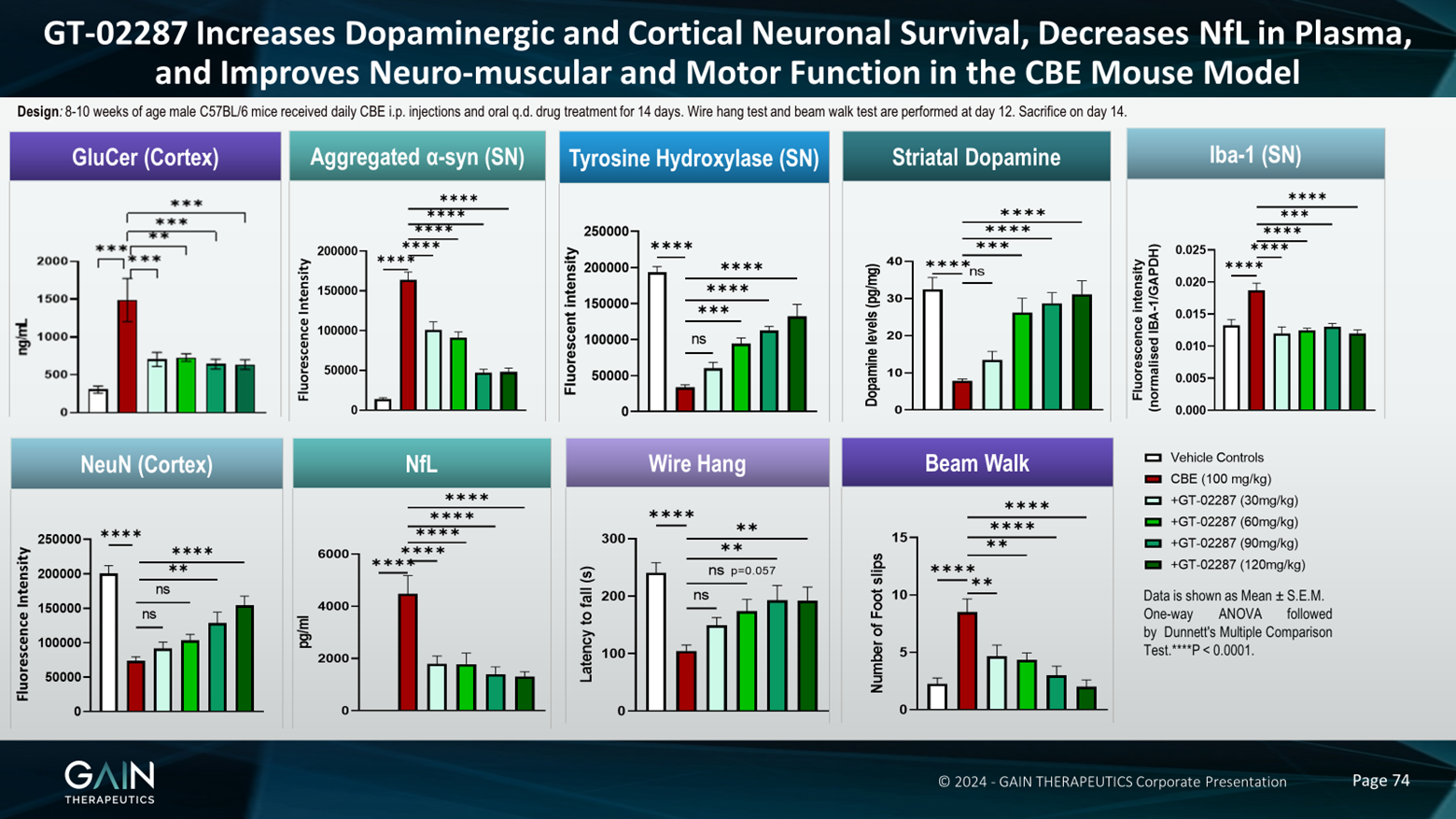
So I'd like to present to you some data in a model which we've used a lot to test this compound and also follow on compounds. It's a model where we've tried to model GBA1 Parkinson's disease in normal animals. So the way we've done this is if we used a compound called CBE, which is an irreversible inhibitor of the GCase enzyme, we administer this compound to the animals and this takes out some of the activity of the GCase enzyme, and it's irreversible. So we can forget that GK is in the model, we've taken it out of function, and then we are testing our compound on the effect on the remaining GCase, which hasn't been inhibited by this compound. So we are trying to model the reduction in GCase activity that we see in GBA1 Parkinson's disease.
In-depth Look at GT2287’s Performance in Animal Studies
And what we see in this model, and what we see with our compound is everything that you'd like to see, really, in a model of GBA1 Parkinson's disease. And with our compound, we see everything that you'd like to see with a molecule, which is treating this disease. So, for example, we see an increase in glucosylceramide, which is a toxic lipid substrate, and we're able to prevent this with our molecule. We've heard a lot about aggregated Alpha-synuclein in this meeting, and in our model, we see a big increase in aggregated Alpha-synuclein and we're able to prevent this with our molecule.
Likewise, we see a decrease in the number of dopaminergic neurons. We see this with tyrosine hydroxylase staining. Tyrosine hydroxylase is an enzyme important in the metabolism or the making of dopamine, and we're able to prevent the loss of these dopaminergic neurons. Maybe unsurprisingly, because we're losing dopaminergic neurons, we also lose striatal dopamine, which we can prevent with our molecule.
We see an increase in neuroinflammation. We see this with EB1 staining. It's a marker of microgliosis. Again, we prevent it with our molecules. We're also preventing the loss of neurons in the cortex. The cortex is, of course, also very important in Parkinson's disease and we've heard, we see cognitive decline, especially in GBA1 Parkinson's disease. So it's important that we can rescue these neurons in the cortex as well.
Very exciting. In our model, we see an increase in the neurofilament light chain in the plasma. So neurofilament light chain is emerging as a marker of neurodegeneration. The fact that we can see it in the plasma suggests that this could be a biomarker for the effects of our compound in an accessible body fluid, i.e., the plasma. The brain is not accessible and there are some issues with taking cerebral spinal fluid as well.
Evaluating GT2287’s Effectiveness After Disease Onset
And then just to round this off, as you might expect, given the hypothesis of how this all works, and given all the other effects that we've seen, we're able to rescue the motor deficits that we see in this model. So when we measure the amount of time a mouse can hang on to a wire or an upturned cage lid, when we administer the CBE, the latency to fall, that's the red bar, is very much reduced. And then we bring this back up towards control levels with our compound. And then in a beam walk experiment, which is measuring coordination, so we count the number of foot slips that the animals make, they're much less coordinated in this model and we're able to prevent this with our compound.
Phase 1 Clinical Trials and Future Plans

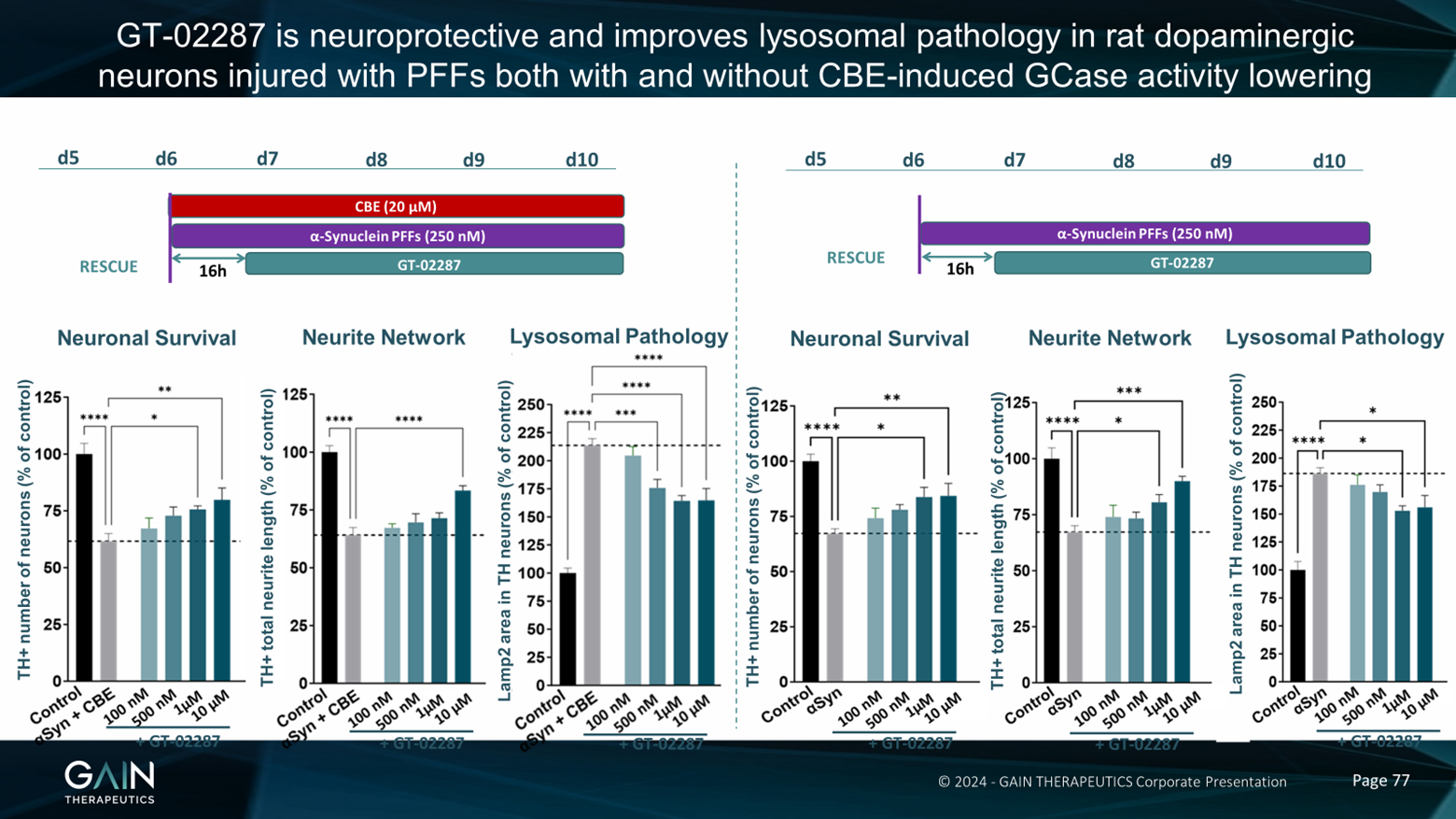
Now, all of the experiments that I just described to you were done by giving our compound at the same time when we introduced the toxic insult. So the really important thing to know is, does our molecule work when the disease process is already underway? So we wanted to take a look at this. And, first of all, we took a look at this in cultured dopamine neurons. So we were looking at the effect of our compound in cultured neurons in a dish. And we've cultured these neurons, and then, as I've just described to you, in the animal model, we've added CBE to these neurons, again, to knock down some of the GCase activity. And then in addition, in this model, we've added Alpha-synuclein-preformed fibrils. So as we heard from Dr. Outeiro when the Alpha-synuclein aggregates together, then this becomes more kind of toxic and pathological, we've added this into the culture as well. And we can see comparing the black control bars and the grey bars, that when we add CBE and Alpha-synuclein, we get a reduction in neuronal survival, and we get a reduction in the neural network. And we also get an increase in lysosomal pathology.
Now, when we add our compound after the start of the experiment, so when all the disease processes are in place, on the next day, essentially, we're still able to rescue these effects on neuronal survival network and lysosome or pathology. What's very interesting, given the discussion that we've just had about idiopathic versus GBA1 Parkinson's disease, is we see very, very similar results when we treat these neurons not with CBE, so we're not affecting the GCase, but when we just add Alpha-synuclein, preformed fibrils, and we see very similar effects in the cell-based model, and we see very similar effects of our compound, i.e., we can rescue this effect and prevent these neurons from dying even in a delayed treatment paradigm.
So we've now taken this into animals, this therapeutic paradigm. And again, we've given the animal CBE, but this time quite a low amount of CBE; half the dose that we gave in the previous in vivo experiment I described to you and we're only giving it every other day. And as we have done with the cell culture experiments, we've also administered Alpha-synuclein, performed the fibrils and we've injected these straight into the brain. Again, to mimic what happens in Parkinson's disease and specifically in this experiment GBA1 Parkinson's disease.
And then once we've got this whole disease cascade going, we started to treat these animals with our compound after either a four-day delay, or an eight-day delay, and then we measured the motor function of these animals in a wire hang test, which I described to you before. So we tested the animals on day 14 of the experiment, which is about a week after we started treating with the compound, and on day 27 of the experiment, which is about three weeks after we started treating with the compound. Then again, we did the motor performance, and the wire hang, and then we also measured plasma NFL as a measure of neurodegeneration.
Conclusion and Acknowledgments
So as I mentioned, this compound is already in the clinic. It's in a Phase 1 trial in healthy volunteers. The upcoming milestones for this trial are this quarter, we expect to complete the single ascending dose part of the phase one trial and to start the multiple ascending dose part of this trial. In the coming quarter, we expect to finish the med part of the trial and we also expect to be able to collect some data looking for effects on GCase activity and levels even in these healthy volunteers. I may not have mentioned but I should have mentioned that our compound seems to act on non-mutated GCase in terms of helping it get to the lysosome and increasing activity as it does in mutated GCase. So we want to look at GCase activity and levels in healthy volunteers as a first inhuman proof of concept.
And then in the second half of this year, spanning out to the first half of next year, we want to add a patient cohort onto the Phase 1 trial, where we will start to look at biomarkers that you can measure then in patients. So, for example, neurofilament light chain, given what we've seen in our preclinical models and other markers of, say, for example, neuroinflammation and so on, and how these lipid substrates are moving as well. And then later on, we expect to have a US IND filing, and then to start our Phase 1B/Phase 2 clinical trial.
So I don't have time for the conclusions, actually, but I think we can go to the question and answer session. But just before I finish, I'd like to point out that I didn't do all this work and it's been carried out by past and present members of the Gain team over the years. I would particularly like to call out Dr Beatrice Guzman, who has produced all of the in vivo data that I shared with you today, and Dr Natalia Perez, who provided the cell-based data.